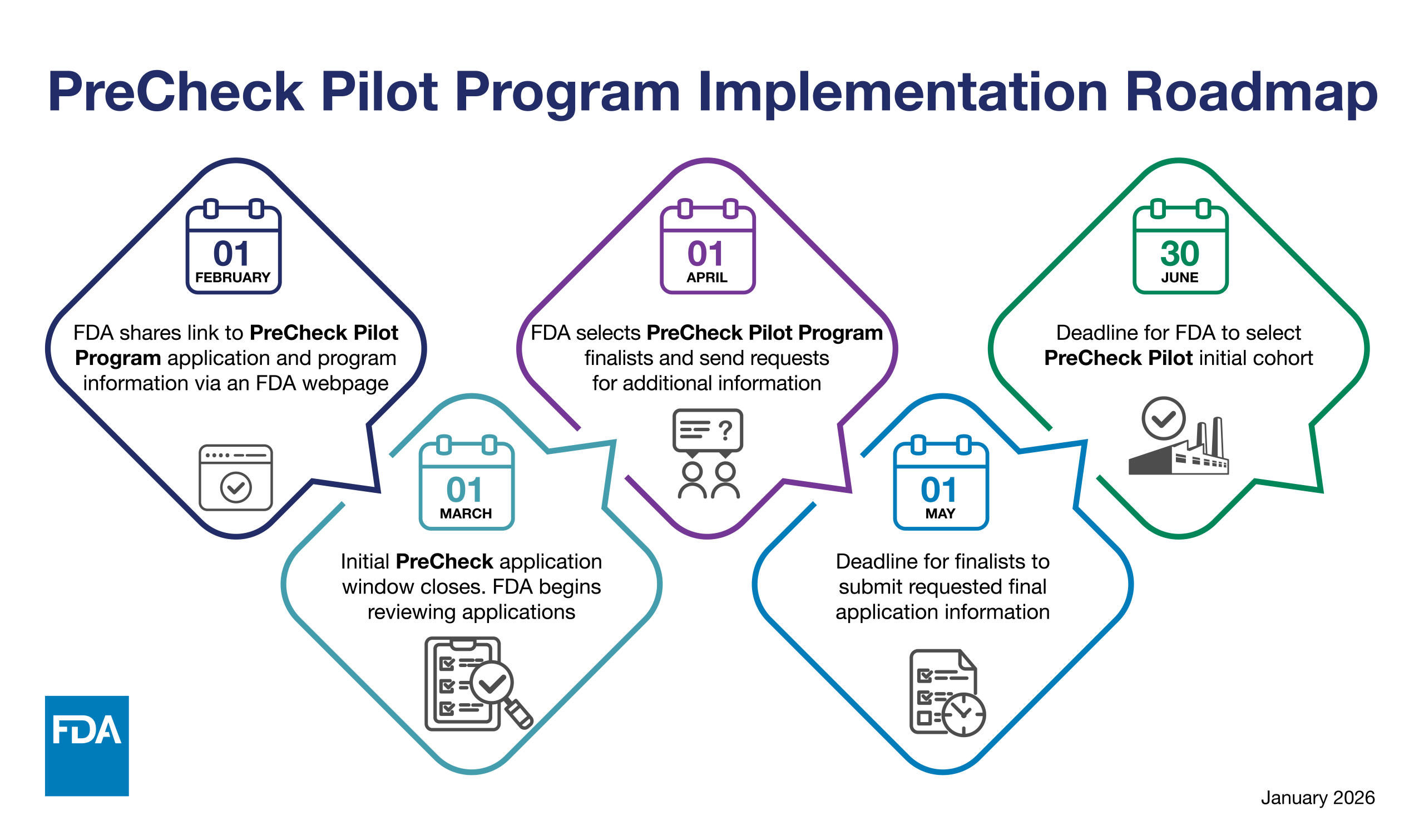
The U.S. Food and Drug Administration announced that it will begin accepting applications on February 1, 2026, for the FDA PreCheck Pilot Program, a transformative initiative designed to strengthen the domestic pharmaceutical supply chain by increasing regulatory predictability and accelerating the development of new manufacturing facilities in the United States.
The PreCheck program represents the FDA’s response to Executive Order 14293, “Regulatory Relief to Promote Domestic Production of Critical Medicines,” and comes at a critical moment when the pharmaceutical industry faces mounting pressure to reshore manufacturing operations. The agency will accept applications through March 1, 2026, with an initial cohort of pilot participants to be selected by June 30, 2026.
Addressing a Critical Supply Chain Vulnerability
The PreCheck program directly addresses a longstanding challenge in U.S. pharmaceutical supply security: more than half of all pharmaceuticals distributed in the U.S. market are manufactured overseas, and the nation remains heavily reliant on international sources for active pharmaceutical ingredients (APIs). Only 11% of manufacturers producing APIs used in FDA-approved products operate within the United States, creating both regulatory complexity and supply chain fragility.
“The PreCheck program will create a more predictable regulatory pathway, accelerate domestic pharmaceutical manufacturing, and protect patient safety,” the FDA stated in announcing the implementation roadmap. The initiative directly responds to industry feedback and stakeholder input gathered during the FDA’s September 2025 public meeting on onshoring manufacturing, where the agency found overwhelming support for early regulatory engagement during facility development phases.
A Two-Phase Regulatory Framework
The PreCheck program introduces an innovative two-phase approach designed to de-risk manufacturing facility development and streamline regulatory review:
Phase 1 – Facility Readiness: This phase establishes a critical engagement window during facility design, construction, and pre-production stages. Manufacturers will receive more frequent FDA communication and early assessment of whether their planned facility and operations, as designed, are likely to comply with current Good Manufacturing Practice (cGMP) requirements. A cornerstone of this phase is the Type V Drug Master File (DMF), a facility-specific dossier that captures comprehensive information including site operations layout, Pharmaceutical Quality System (PQS) elements, and Quality Management Maturity practices. This facility-focused DMF can subsequently be incorporated by reference into individual product applications, eliminating redundant regulatory reviews.
Phase 2 – Application Submission: This phase focuses on streamlining the Chemistry, Manufacturing, and Controls (CMC) section of new drug applications and biologics license applications (BLAs). Manufacturers receive pre-application meetings, early CMC feedback, and frontloaded assessment activities that align inspection schedules with review cycles—accelerating the overall regulatory timeline.
Selection Criteria Align with Strategic Priorities
The FDA will select participants based on their overall alignment with national priorities, including the type of products to be manufactured, the phase of facility development, the timeline for producing pharmaceuticals for the U.S. market, and the degree of innovation in facility development. This selective approach ensures that pilot resources are concentrated on facilities most likely to address supply chain vulnerabilities in critical therapeutic areas.
Industry Response: Unprecedented Investment Momentum
The announcement arrives amid an unprecedented wave of manufacturing investment commitments. According to recent industry tracking, pharmaceutical companies have committed more than $480 billion toward manufacturing and research and development projects in the United States as of November 2025, with plans to establish 22 new manufacturing sites and generate approximately 44,000 new jobs. Major pharmaceutical companies—including Eli Lilly, Johnson & Johnson, Merck, Pfizer, Novo Nordisk, AstraZeneca, and others—have announced ambitious reshoring initiatives spanning APIs, sterile injectables, biologics, and specialized modalities including radioligand therapies and cell and gene therapies.
Eli Lilly’s $27 billion commitment to expand domestic manufacturing of APIs and sterile injectables exemplifies the scale and scope of current reshoring activity, which accelerated following the Trump administration’s April 2025 “Liberation Day” tariff announcements. The PreCheck program is expected to substantially reduce barriers to translating these announced commitments into operational manufacturing capacity.
Addressing Industry Concerns and Incorporating Stakeholder Feedback
The FDA’s implementation roadmap reflects extensive stakeholder consultation conducted during 2025. Industry respondents previously expressed concerns about the ambiguity and cost implications of facility-related regulatory delays, where isolated facility compliance issues could trigger drug application rejections. The PreCheck program directly addresses this risk by enabling proactive facility assessment before product submissions are filed.
Stakeholders also emphasized the importance of early FDA engagement during facility planning and construction phases—a point the FDA has explicitly incorporated. “Overall sentiment regarding the PreCheck program was positive, with industry requesting early engagement during facility development phases,” the agency noted.
However, details remain pending. The FDA has indicated that comprehensive program guidelines, application content requirements, evaluation criteria, and post-selection timelines will be released on February 1, 2026, when the application portal goes live. Industry observers expect the additional guidance to clarify expectations for the Type V DMF architecture, engagement protocols, and the anticipated benefits of pilot participation.
Market Context: Manufacturing Capacity and Cost Constraints
While regulatory predictability is critical, industry analysts note that manufacturing reshoring faces countervailing economic headwinds. Despite PreCheck’s potential to accelerate facility approvals, U.S. manufacturing costs remain substantially higher than overseas alternatives, creating sustained competitive pressure. Global pharmaceutical production surged 9.1% in 2025 primarily due to front-loading activity in anticipation of tariffs, with industry observers expecting production growth to moderate to 1.6% in 2026 as supply chain normalization occurs.
The PreCheck program complements but does not eliminate these structural cost challenges. Analysts project that high U.S. production costs could continue to favor offshore manufacturing for non-critical commodities, even as tariff threats and supply chain resilience imperatives drive nearshoring of high-priority, national security-critical products.
Operational Excellence and Capacity Bottlenecks
Stakeholders have also emphasized that regulatory streamlining must be paired with attention to broader manufacturing bottlenecks. Contract development and manufacturing organizations (CDMOs) report tightening capacity for biologics and fill-finish operations, with timelines becoming increasingly constrained. Specialized unit operations—such as micronization under containment and advanced aseptic processing—face particular capacity limitations, suggesting that PreCheck’s regulatory benefits may intersect with CDMO availability constraints.
Strategic Implications for the Pharmaceutical Industry
The PreCheck program represents a pivotal policy shift toward supporting domestic pharmaceutical manufacturing through regulatory efficiency rather than capital subsidies or direct incentives. By reducing the perceived regulatory risk and timeline uncertainty associated with new facility development, the program aims to unlock private sector investment and accelerate the translation of announced reshoring commitments into operational capacity.
For manufacturers considering U.S. facility investments, PreCheck participation offers material advantages: earlier regulatory alignment, reduced risk of facility-related application delays, and the creation of reusable facility-specific regulatory documentation through the Type V DMF mechanism. These benefits could prove decisive, particularly for mid-sized manufacturers and specialized producers considering U.S. investments in high-margin or supply-critical therapeutic categories.
For the FDA, the program represents a strategic reframing of its role from post-hoc facility inspector to proactive regulatory partner, providing early technical guidance during facility design phases when course corrections remain feasible and cost-effective.
Next Steps and Application Timeline
Industry participants interested in the PreCheck Pilot Program should prepare applications for submission between February 1 and March 1, 2026. The FDA will announce the initial cohort of selected facilities by June 30, 2026. Comprehensive program details, including application guidelines, evaluation criteria, and facility engagement protocols, will be available on the FDA’s PreCheck application website on February 1, 2026.
